Differentiation of Gene Richness on Duplicated Chromosomes and Survey of Genes Captured by ESTs in Poplar Genome 
 Author
Author  Correspondence author
Correspondence author
Genomics and Applied Biology, 2010, Vol. 1, No. 3 doi: 10.5376/gab.2010.01.0003
Received: 16 Oct., 2010 Accepted: 09 Nov., 2010 Published: 29 Dec., 2010
This article was first published in Genomics and Applied Biology (Regular Print Version), and here was authorized to redistribute under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Li et al., 2010, Differentiation of Gene Richness on Duplicated Chromosomes and Survey of Genes Captured by ESTs in Poplar Genome, Genomics and Applied Biology, 29(3): 570-576 (DOI: 10.3969/gab.029.000570)
In higher plant genomes, most sequences are unexpression sequence, the proportion of a genome that encodes for genes may be very small. Understanding gene distribution in the genome is a very important aspect for invastigating the genome structure. Funded by the U.S. Department of Energy, a clonal Populus trichocarpa genome sequencing has been completed and released to the public. The accomplishment of poplar genome offers us a unique opportunity to survey the gene distribution in the genome of a forest tree. In this paper, based on Poisson calculator, we investigated the gene density of various chromosomes in poplar genome. As a result, we found that gene density is significantly different among chromosomes in poplar genome. Modern poplar genome arose from an ancient whole genome duplication event, known as “salicoid duplication”. Thus poplar genome shared large duplication segments among different chromosome members. However, our results demonstrated that gene abundance pattern was different from the chromosomal duplication pattern of poplar genome. This implied the duplicated genes lost at different rates on the duplicated chromosomes following the salicoid duplication. Meanwhile, based on alignment with about 90 thousands ESTs,we found that only 16.8% of the predicted gene models in poplar genome had EST proofs. Although, EST sequencing is an attractive alternative to whole genome sequencing for gene identification, the power of small scale EST sequencing study should be properly evaluated since with limited number of EST sequences, genes captured by ESTs are fairly limited.
Forests are the principal representatives of the terrestrial ecosystem and provide important sustainable resources for humankind (Li and Yin, 2007). Although forests only cover about 30% of the earth’s land surface, they allocated over 80% of CO2 from the atmosphere through green vegetations. Thus, increasing forest productivity is also an essential approach to reduce the greenhouse gas. However, our understanding for forest trees is limited in comparison with that for many other organisms, and better understanding of the genetic mechanisms influencing tree adaptation and productivity is important for the management of the world’s forest resources (Caetano-Anollés and Gresshoff, 1997). The genus Populus possesses many characteristics that are conductive to functional genomics, which leads the emergency of poplar as the model system for tree genome research (Wullschleger et al., 2002). Under the efforts of numerous scientists worldwide, the genome of a black cottonwood (Populus trichocarpa Torr. & Gray ex Brayshaw), “clone 383-2499”, has been sequenced and publicly released (Tuskan et al., 2006). It is the first sequence of a woody perennial plant.
In recent decades, sequencing capacity available to biological scientist increases in an exponential manner. Several plant genomes have been sequenced (Lyons and Freeling, 2008). However, whole genome sequences for most plant species are still unavailable in the near future, especially for tree species. The genome sequence of poplar offers us a unique opportunity to learn the genome characteristics of a tree species. In the earlier studies, scientists have discovered many interesting findings, for example, the poplar genome project revealed that the chromosomes of modern Populus arose from an ancient whole-genome duplication event known as “salicoid duplication”, and the 19 haploid chromosomes in poplar genome might evolve from 10 ancestral chromosomes (Tuskan et al., 2006). Meanwhile, the comparative mapping project demonstrated that the genomes of different poplar species maintained the basic genome structure formed by the salicoid duplication event (Yin et al., 2008). Compare to these findings, gene distribution is also an important aspect to explore the characteristics of poplar genome. Relating to this point, there was only a short descriptive paragraph in the poplar genome paper, but no statistical analyses were conducted in that article (Tuskan et al., 2006). In this paper, the Poisson calculator is employed to survey gene density on different chromosomes of poplar genome. In this study, we especially address on two interesting questions: first, do the duplicated chromosomes have the similar gene density? Second, how many percent of genes in poplar genome can be captured by a moderate scale EST sequencing project?
1 Results
1.1 Compare gene densities between chromosomes that share large duplicated segments
Based on the Poisson calculator, genes were found to occur at different frequency on different chromosome members. Six chromosomes, including chromosome II, VI, VIII, IX, X, and XV, were found to be overabundant with genes; eight of them, including chromosome I, IV, XI, XII, XIII, XVII, XVIII, and XIX, were sparse with genes; only four chromosomes, including chromosome III, V, XIV, and XVI, had gene quantities that didn’t significantly apart from the expected numbers (table 1). Therefore, chromosomes can be classified into three classes based on the gene density: chromosomes with more genes than the expected numbers; chromosomes with genes quantity that do not significantly apart from the expected numbers; chromosomes with less genes than the expected numbers.
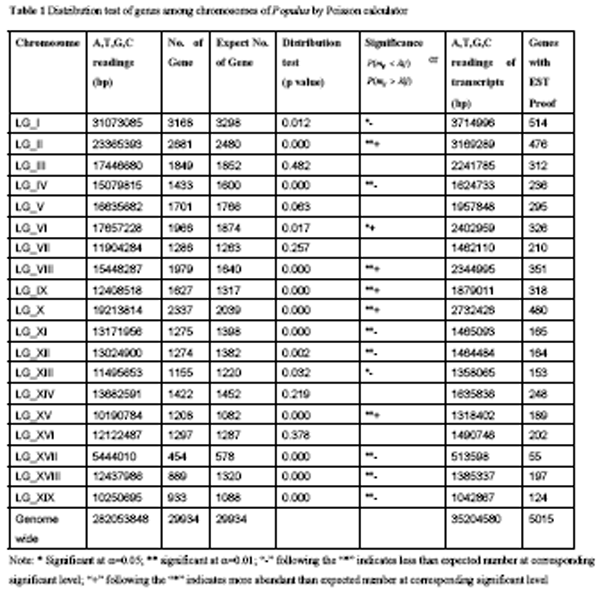 Table 1 Distribution test of genes among chromosomes of Populus by Poisson calculator |
Visual plotting of gene distribution within each chromosome and homology among chromosome members of poplar genome were displayed in figure 1. From this figure, we found that genes distributed relatively evenly within each chromosome. We didn’t detect regions with low gene density that might correspond to the centromeric and telomeric regions. Since the current sequence scaffolds mapped onto different chromosomes only account 86% of the total poplar genome and repetitive sequences tend to be more difficult to be assembled in the shotgun sequencing project, we proposed these regions might embed in the sequence scaffolds that hadn’t mapped onto chromosomes.
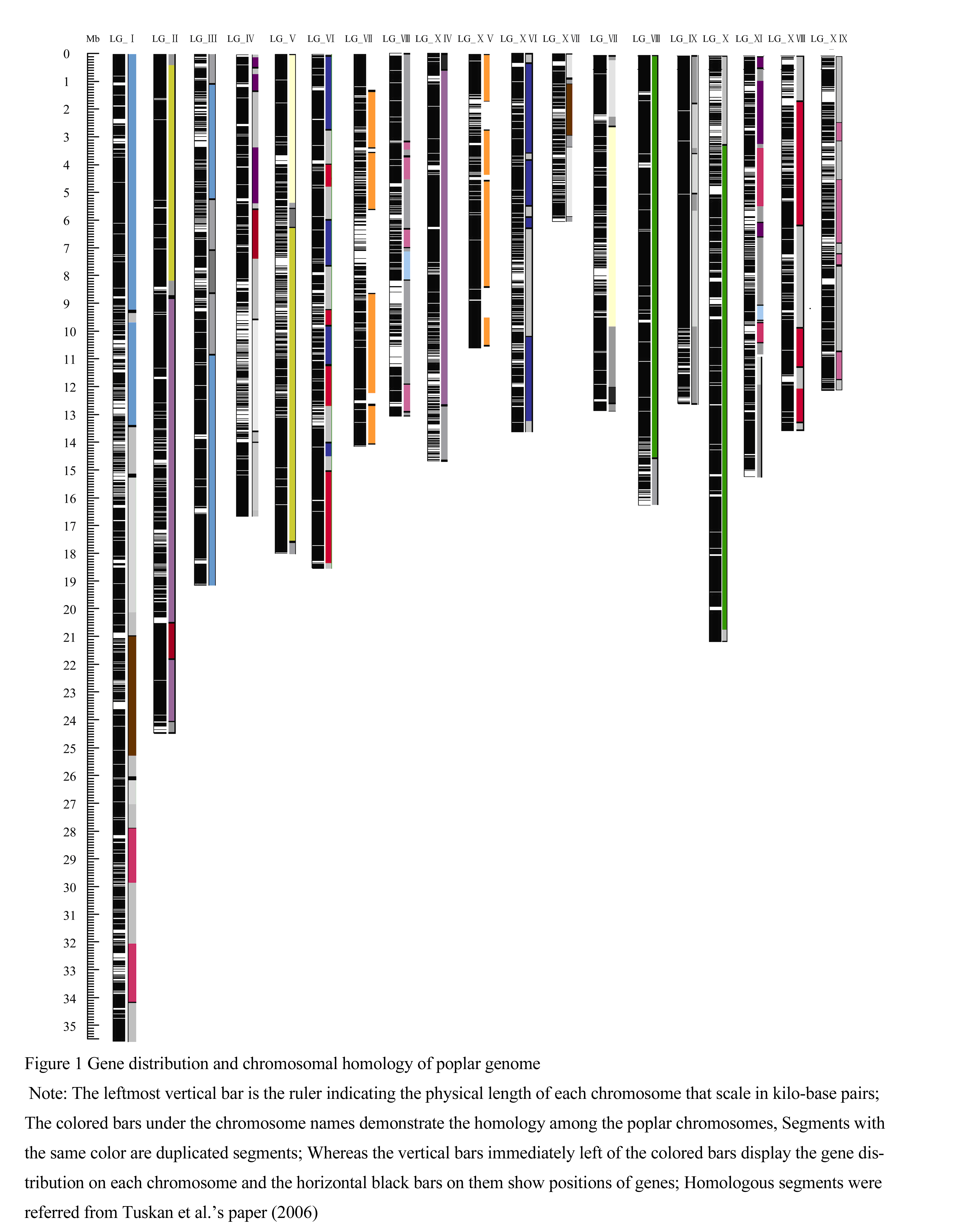 Figure 1 Gene distribution and chromosomal homology of poplar genome |
Referring to the poplar genome paper (Tuskan et al., 2006), the 19 haploid chromosomes of poplar genome were duplicated from 10 ancestral chromosomes. However, the pattern of chromosomes overabundant, even, or sparse with genes is different from the chromosomal homologous pattern as revealed by Tuskan and his colleagues (Tuskan et al., 2006). We compare eight pairs of chromosomes that share large duplication segments (table 2) and find that only gene densities on two pairs of the chromosomes are in the same class, whereas gene densities on the other six homologous pairs of chromosomes are in different classes. For example, chromosome XII and XV are in high homology, but their gene densities are in opposite classes. Chromosome XII is overabundant with genes. By contrast, chromosome XV is sparse with genes.
.png) Table 2 Comparison of gene densities on the chromosomes that share duplication segments |
1.2 Survey gene models captured by ESTs
Genome can be classified into intergenic and genic regions. Genic region is consisted of 5’UTR, exons, introns, and 3’ UTR and the regulatory elements. When a gene is transcribed into mature mRNA, its introns were splice off. Gene sequences that can be captured by EST sequencing would include the 5’ UTR, exons, and 3’ UTR. In this study, we total investigate 29 934 putative genes. The average length of these genes is 2 690 bp, and the median length is 1 951 bp, indicating the significant skewness to genes in short lengths. The minimum length of gene is 149 bp, whereas the largest gene covers 34 109 bp. Further analysis revealed that, in poplars, the average exon lengths of genes are 1 176 bp, coding 392 amino acids. From this analysis, we conclude that, on average, exon sequences account 43.7% of the gene sequences. The total length of transcripts (sequences covered by mRNAs) were found to only account 12.5% of the valid A,T,G,C readings of the assembled genome sequences. There are about 90 thousand EST sequences of P. trichocarpa deposite in GeneBank. We summarize the gene models captured by these ESTs and found that they only covered about 16.8% of the total genes in poplar genome. The poor gene coverage might relate to that EST sequences depend a lot on the tissue library and normally contain heavily redundant sequences (Susko and Roger, 2004; Wang et al., 2005).
2 Discussion
Most eukaryotic genomes have numerous duplicated genes, many of which appear to have arisen from one or more cycles of ancient polyploidy (paleopolyploidy) (Adams and Wendel, 2005; Adams et al., 2004; Blanc and Wolfe, 2004). Following paleopolyploidy (genome doubling), there is extensive loss of duplicated genes (Adams and Wendel, 2005; Adams et al., 2004; Blanc and Wolfe, 2004). Cytological studies revealed that almost all Populus existed in the diploid form with a haploid number of chromosomes equal to 19 (Smith, 1943). However, poplar genome sequencing project revealed that the modern poplar genome arose from an ancient whole genome duplication event, known as “salicoid duplication”. Thus polar chromosomes share high sequence homology (Tuskan et al., 2006). However, the pattern of chromosomes overabundant, even, or sparse with genes is different from the homologous pattern among chromosomes as revealed by the poplar genome sequencing project (Tuskan et al., 2006). This suggests the loss of genes occurs at different rate on chromosomes that share large duplicated segments after the salicoid duplication.
ESTs are generated by partially sequencing randomly isolated gene transcripts that have been converted into cDNA (Adams et al., 1991). EST sequencing has played an important role in the identification, discovery and characterization of organisms as they provide an attractive and efficient alternative to whole genome sequencing (Lijoi et al., 2007). Concerted, high budget EST sequencing projects have been carried out for many of the economically and ecologically important plant species. In high plants, the majority of their genome sequences are consisted of noncoding sequences. Gene sequences only account small parts of their genomes. In poplar, the total transcript sequences only account for 12.5% of the genome sequences. Alignment analysis of the EST sequences from P. trichocarpa deposited in GenBank revealed that over 100 thousand ESTs only cover 16.8% of the putative gene models annotated in the poplar Vista browser. In high organisms, such as mammalian and Arabidopsis, even concerted, high-budget EST sequencing can only get about 50%~60% of genes (Pers. Comm.). Thus, genes captured by limited number of ESTs are very incomplete. This is because that, first, ESTs sequences are heavily redundant; second, EST sequences are tissue type dependent, in a particular tissue library, some genes have high expression level and these genes would be sequenced repetitively, but there are many other genes with little chance to be sequenced. Although EST sequencing is an attractive alternative to whole genome sequencing for gene identification, the power of small scale EST sequencing studies should be properly evaluated since with limited effort and budget, genes captured by ESTs are fairly limited.
In our analysis, we used gene models from the JGI annotation database in Vista browser. These gene models showed high conservation with the Arabidopsis gene sets and are most likely “real” genes. With percentage of unreliable annotations in the current Jamboree models, we believe that it was better to focus on conserved real genes. In our analysis we included nearly 30 000 gene models (those that most likely real genes) which were from the unambiguously mapped sequence scaffolds along 19 chromosomes of poplar genome (about 86% of the total genome length). Although we didn’t cover all of the genes and sequences of poplar genome, the number of genes and genomic sequences included in this study should be sufficient to give us a general idea to answer the questions addressed in this paper.
3 Materials and Methods
The genome sequence and gene information is obtained from poplar genome browser (http://shake.jgi-psf.org/ cgi-in/searchGM?db=Poptr1_1). Gene distribution among chromosome members was evaluated by the observed number of genes comparing with their expectations under the Poisson distribution. The expected gene number in chromosome would be a sample from a Poisson distribution, λi=mLi/Σ<sub>i</sub>Li , where is the total number of genes detected within chromosome ; is the length of A,T,C,G reading of chromosome . The probabilities and were evaluated under the cumulative Poisson distribution at =0.05 and =0.01 significant level. Visual plotting of gene distribution on each chromosome is drawn by the Mapchart software (Voorrips, 2002). Chromosome homology was referred to that revealed by Tuskan et al. (2006). P. trichocarpa EST seuquences were from Genbank, and EST coverage was summarized from the gene models predicated in the Vista browser (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html).
Authors' contributions
Conceived the experiment and edited the manuscript: Tongming Yin. Data collection and analysis: Shuxian Li, Xiaogang Dai and Handong Gao. Wrote the manuscript: Shuxian Li. All authors have read the final version of this paper and agreed with the authors’ credits.
Acknowledgments
Funding for this research was provided by the Natural Science Foundation of China (30971609) and Forestry Nonprofit Project (200904002). The authors would like to thank for the scientists who maintain the website (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.home.html) and make the poplar sequences available to the public. We also thank for Prof. Mingxiu Wang for his critical insights during the writing and revision of this manuscript. Special thanks go to the editor and anonymous reviewers for their help in formulating the final revision.
References
Adams M.D., Kelley J.M., Gocayne J.D., Dubnick M., Polymeropoulos M.H., Xiao H., Merril C.R., Wu A., Olde B., Moreno R.F., Kerlavage A.R., McCombie W.R., and Venter J.C., 1991, Complementary DNA sequencing: Expressed sequence tags and human genome project, Science, 252: 1651-1656 doi:10.1126/science.2047873 PMid:2047873
Adams K.L., Percifield R., and Wendel J.F., 2004, Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid, Genetics, 168(4): 2217-2226 doi:10.1534/genetics.104.033522 PMid:15371349 PMCid:1448729
Adams K.L., and Wendel J.F., 2005, Novel patterns of gene expression in polyploid plants, Trends in Genetics, 21(10): 539-543 doi:10.1016/j.tig.2005.07.009 PMid:16098633
Blanc G., and Wolfe K.H., 2004, Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution, Plant Cell, 16(7): 1679-1691 doi:10.1105/tpc.021410 PMid:15208398 PMCid:514153
Caetano-Anollés G., and Gresshoff P.M., eds, 1997, DNA markers: protocols, applications and overviews, Wiley-VCH, New York, pp.1-32
Li S.X., and Yin T.M., 2007, Map and analysis of microsatellites in genome of Populus: The first sequenced perennial plant, Science in China. Series C, Life sciences, 50(5): 690-699 doi:10.1007/s11427-007-0073-6 PMid:17879069
Lijoi A., Mena R.H., and Prünster I., 2007, A bayesian nonparametric method for prediction in EST analysis, BMC Bioinformatics, 8:339 doi:10.1186/1471-2105-8-339 PMid:17868445 PMCid:2220008
Lyons E., and Freeling M., 2008, How to usefully compare homologous plant genes and chromosomes as DNA sequences, Plant J., 53(1): 661-673 doi:10.1111/j.1365-313X.2007.03326.x PMid:18269575
Smith E.C., 1943, A study of cytology and speciation in the genus Populus L., J. Arnold Arbor, 24: 275-305
Susko E., and Roger A.J., 2004, Estimating and comparing the rates of gene discovery and expressed sequence tag (EST) frequencies in EST surveys, Bioinformatics, 20(14): 2279-2287 doi:10.1093/bioinformatics/bth239 PMid:15059814
Tuskan G.A., DiFazio S., Jansson S., Bohlmann J., Grigoriev I., Hellsten U., Putnam N., Ralph S., Rombauts S., Salamov A., Schein J., Sterck L., Aerts A., Bhalerao R.R., Bhalerao R.P., Blaudez D., Boerjan W., Brun A., Brunner A., Busov V., Campbell M., Carlson J., Chalot M., Chapman J., Chen G.L., Cooper D., Coutinho P.M., Couturier J., Covert S., Cronk Q., Cunningham R., Davis J., Degroeve S., Déjardin A., dePamphilis C., Detter J., Dirks B., Dubchak I., Duplessis S., Ehlting J., Ellis B., Gendler K., Goodstein D., Gribskov M., Grimwood J., Groover A., Gunter L., Hamberger B., Heinze B., Helariutta Y., Henrissat B., Holligan D., Holt R., Huang W., Islam-Faridi N., Jones S., Jones-Rhoades M., Jorgensen R., Joshi C., Kangasjärvi J., Karlsson J., Kelleher C., Kirkpatrick R., Kirst M., Kohler A., Kalluri U., Larimer F., Leebens-Mack J., Leplé J.C., Locascio P., Lou Y., Lucas S., Martin F., Montanini B., Napoli C., Nelson D.R., Nelson C., Nieminen K., Nilsson O., Pereda V., Peter G., Philippe R., Pilate G., Poliakov A., Razumovskaya J., Richardson P., Rinaldi C., Ritland K., Rouzé P., Ryaboy D., Schmutz J., Schrader J., Segerman B., Shin H., Siddiqui A., Sterky F., Terry A., Tsai C.J., Uberbacher E., Unneberg P., Vahala J., Wall K., Wessler S., Yang G., Yin T., Douglas C., Marra M., Sandberg G., Van de Peer Y., and Rokhsar D., 2006, The genome of black cottonwood, Populus trichocarpa (Torr.& Gray), Science, 313(5793): 1596-1604 doi:10.1126/science.1128691 PMid:16973872
Voorrips R.E., 2002, MapChart: software for the graphical presentation of linkage maps and QTLs, J. Hered, 93(1): 77-78 doi:10.1093/jhered/93.1.77 PMid:12011185
Wang J.P.Z., Lindsay B.G., Cui L.Y., Wall P.K., Marion J., Zhang J.X., and dePamphilis C.W., 2005, Gene capture prediction and overlap estimation in EST sequencing from one or multiple libraries, BMC Bioinformatics, 6: 300 doi:10.1186/1471-2105-6-300 PMid:16351717 PMCid:1369009
Wullschleger S.D., Jansson S., and Taylor G., 2002, Genomics and forest biology: Populus emerges as the perennial favorite, The Plant Cell, 14: 2651-2655 doi:10.1105/tpc.141120 PMCid:540295
Yin T.M., DiFazio S.P., Gunter L.E., Zhang X.Y., Sewell M.M., Woolbright S.A., Allan G.J., Kelleher C.T., Douglas C.J., Wang M.X., and Tuskan G.A., 2008, Genome structure and emerging evidence of an incipient sex chromosome in Populus, Genome Res., 18(3): 422-430 doi:10.1101/gr.7076308 PMid:18256239 PMCid:2259106



. PDF(260KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Shuxian Li
. Xiaogang Dai
. Handong Gao
. Tongming Yin
Related articles
. Poplar genome
. Gene distribution
. EST coverage
. Salicoid duplication event
Tools
. Email to a friend
. Post a comment