Research Article
Polygenic Risk Scores (PRS/PGS) across Multi-ancestry and Cross-domain Settings: Statistical Framework, Methodological Advances, and Robustness Evaluation 
 Author
Author  Correspondence author
Correspondence author
Genomics and Applied Biology, 2026, Vol. 17, No. 3
Received: 06 Apr., 2026 Accepted: 12 May, 2026 Published: 25 May, 2026
Polygenic risk scores (PRS/PGS) aggregate genome-wide association study (GWAS) effect sizes to quantify individual-level genetic susceptibility, serving as a key bridge between genetic association findings and practical applications. With the rapid expansion of large-scale genotype-phenotype datasets, PRS methodology has evolved from early clumping-and-thresholding (C+T) approaches to frameworks that explicitly model linkage disequilibrium (LD) and effect size distributions using Bayesian shrinkage and penalized regression, and further incorporate functional annotations, multi-ancestry data, and transfer learning to improve predictive performance and interpretability. However, the portability and robustness of PRS across populations remain major challenges, often manifesting as reduced predictive accuracy, calibration bias, and unstable decision thresholds. From a statistical perspective, these issues can be understood as an estimand mismatch arising from differences in LD structure, allele frequency spectra, and effect distributions across populations. In this study, we revisit PRS within a unified statistical genetics framework by conceptualizing it as a model-dependent predictive functional, and link it to SNP heritability as part of a continuous inference chain from variance decomposition to individual-level prediction. Building on this perspective, we systematically review and compare state-of-the-art PRS methods under multi-population settings, including LD-aware Bayesian shrinkage, functionally informed models, multi-ancestry transfer learning, and model stacking and recalibration strategies, with representative methods such as PRS-CSx, CT-SLEB, and PolyPred. We further propose a standardized analytical workflow of “training-validation-freezing-external evaluation” and advocate a multi-dimensional evaluation framework based on relative R²/AUC, calibration metrics, and decision-curve net benefit. In addition, we discuss joint modeling of PRS with environmental and lifestyle factors and its applications in both human health and crop breeding. Finally, we address issues of cross-population inequity and ethical governance, and propose an integrated framework centered on multi-ancestry data expansion, causal and functional annotation integration, ancestry-aware modeling, environment coupling, and population-specific recalibration. This framework aims to promote PRS/PGS from a predictive tool toward a transferable, interpretable, and equitable decision-support system, providing a systematic foundation for the application of complex trait genetics.
Polygenic risk scores (PRS/PGS) aggregate effect size estimates derived from genome-wide association studies (GWAS) to generate individual-level predictions, thereby transforming locus-trait associations into quantitative measures of genetic susceptibility for complex traits or diseases. With the continuous expansion of large-scale genotype-phenotype cohorts and advances in computational methods, PRS construction has evolved from early clumping-and-thresholding (C+T) approaches to frameworks that explicitly model linkage disequilibrium (LD) and effect size sparsity using Bayesian shrinkage and penalized regression. More recently, these methods have further incorporated functional annotations, fine-mapping, and multi-trait information to enhance signal-to-noise ratio, interpretability, and predictive performance. This methodological progression reflects a paradigm shift in statistical genetics from hypothesis-driven analyses to genome-wide, hypothesis-free scanning (Cai et al., 2021; Weissbrod et al., 2022; Zhang et al., 2023; Fang and Wu, 2026). Meanwhile, multi-ancestry training and cross-population transfer learning approaches have rapidly developed, and the increasing availability of open-access GWAS summary statistics and LD reference panels has facilitated reproducibility, cross-platform application, and secondary analyses (Wang et al., 2023).
Across both human medicine and crop breeding, PRS/PGS share a fundamentally analogous objective: to enable early prediction, risk stratification, and selection decisions at the individual level under resource constraints. In medical research, PRS provides a relatively stable estimate of genetic risk across the life course beyond traditional risk factors, supporting stratified screening, longitudinal monitoring, and personalized intervention (Lennon et al., 2024; Xiang et al., 2024). In crop breeding, PGS is methodologically aligned with genomic selection, and is particularly valuable for traits that are costly or late to measure (e.g., perennial crops or complex stress-related traits), where it can serve as an early surrogate phenotype for individual selection, parental optimization, and cross prediction, thereby increasing genetic gain per unit time or cost (Sima et al., 2024). Consequently, establishing a unified methodological language and evaluation framework across medicine and breeding has become an important direction for advancing the application of statistical genetics.
Despite continuous methodological and data advances, the portability and robustness of PRS/PGS across populations remain major challenges. Predictive performance is highly dependent on the allele frequency spectrum and LD structure of the training population. When LD patterns and the tagging relationships between SNPs and causal variants differ across ancestries, signal attenuation or effect size distortion commonly occurs during extrapolation. In addition, population-specific effects, gene-environment interactions, and heterogeneity in phenotype definition and measurement further contribute to reduced explanatory power, calibration bias, and instability of decision thresholds. From a statistical inference perspective, these issues can be understood as an estimand mismatch arising from differences in LD structure, allele frequency spectra, and effect distributions across populations (Duncan et al., 2019; Jayasinghe et al., 2024; Fang, 2026). This structural bias manifests consistently across both human and agricultural systems and extends to concerns regarding population fairness and practical implementation. Moreover, imbalances in sample size and resource availability across ancestries exacerbate the underperformance of PRS in underrepresented populations, making cross-population PRS applications a complex challenge involving statistical modeling, data infrastructure, and ethical governance.
Under this context, it is necessary to re-examine the nature of PRS/PGS from a statistical inference perspective. Strictly speaking, PRS is not a direct estimate of “true genetic risk,” but rather a model-dependent predictive functional defined by the training data, LD structure, and effect estimation model. This perspective is intrinsically consistent with the statistical interpretation of SNP heritability (Fang, 2026): the latter quantifies the proportion of phenotypic variance explained by additive genetic effects under a given set of markers and model assumptions, whereas PRS projects this genetic signal into an individual-level predictive quantity under the same informational constraints. In other words, SNP heritability reflects variance explained at the population level, whereas PRS reflects predictive ability at the individual level, together forming a continuous inference chain from variance decomposition to individual prediction.
Within this unified framework, differences among PRS methods (e.g., C+T, LD-aware Bayesian shrinkage, and multi-ancestry transfer models) fundamentally correspond to different modeling assumptions regarding effect size distributions, LD structure, and sparsity, thereby implying different statistical targets (estimands). Consequently, PRS performance depends not only on sample size and data quality, but also on the degree of alignment between model assumptions and the target population. The decline in cross-population predictive performance can thus be interpreted as a manifestation of estimand mismatch at the level of individual prediction.
Building on this perspective, the present study focuses on the “individual prediction layer” within the broader framework of statistical genetics, extending prior work on methodological paradigm evolution (Fang and Wu, 2026) and variance-based inference (Fang, 2026). We systematically review and compare state-of-the-art PRS/PGS methods in multi-population contexts, including cross-ancestry effect estimation and transfer learning, ancestry-aware LD modeling, functional annotation and causal refinement, as well as model stacking and recalibration strategies, with representative methods such as PRS-CSx, CT-SLEB, and PolyPred. At the level of evaluation and practice, we propose a standardized workflow of “training-validation-freezing-external evaluation,” emphasizing a multi-dimensional assessment based on relative R²/AUC, calibration slope, and decision-curve net benefit. This framework is further complemented by ancestry-stratified and LD-perturbation sensitivity analyses, small-sample recalibration in target populations, and cost-benefit evaluation, supported by benchmark datasets, reference panels, and transparent reporting standards, with the aim of promoting the development of PRS/PGS as a transferable, interpretable, and equitable tool for applications in human health and crop improvement.
1 Fundamental Framework and Methodological Evolution of PRS/PGS
From a statistical inference perspective, the construction of PRS/PGS can be understood as a process that translates population-level association signals into individual-level predictive quantities. Specifically, the marginal effect estimates obtained from GWAS are not directly suitable for prediction; instead, they must be re-estimated under appropriate modeling assumptions that account for linkage disequilibrium structure and effect size distributions. This typically involves shrinkage and aggregation procedures that yield more stable and generalizable effect representations. These regularized effects are then projected onto individual genotype profiles to quantify genetic risk at the individual level. In this sense, PRS can be viewed as a model-dependent predictive function, jointly determined by effect estimation, regularization strategies, and genotype encoding, reflecting a continuous inferential pathway from association signal extraction to individual risk prediction.
Differences among methods fundamentally arise from distinct modeling assumptions regarding effect size distributions, linkage disequilibrium (LD) structure, and sparsity, thereby implying different statistical targets (estimands). Under this framework, the evolution of PRS methodology can be understood as a progression from “independent locus approximation” to “LD-aware modeling,” and further toward the integration of functional and ancestry information.
1.1 Classical clumping and thresholding (C+T)
Clumping and thresholding (C+T) is one of the earliest and most widely used approaches for constructing PRS/PGS (Sima et al., 2024) (Figure 1). This method begins with single-marker GWAS effect estimates, ranks candidate variants by statistical significance (p-values), and performs LD clumping using predefined window sizes and r² thresholds to retain representative “sentinel” SNPs. Individual scores are then calculated via linear aggregation:
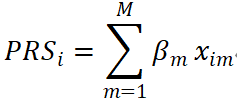

where βm denotes the marginal effect size of the m-th SNP, and xim represents the genotype of individual i.
|
Figure 1 Workflow of the clumping and thresholding (C+T) approach for constructing polygenic risk scores (PRS) Note: This figure illustrates the standard workflow of the clumping and thresholding (C+T) method for constructing polygenic risk scores (PRS). Starting from GWAS summary statistics, single-marker effect estimates (β) are obtained and used for variant selection. SNPs are first ranked by statistical significance (p-values), followed by linkage disequilibrium (LD)-based clumping within a specified genomic window and r2 threshold, retaining representative “sentinel” variants while removing correlated markers. The selected variants are then aggregated into an individual-level PRS using a linear scoring function, where SNP effect sizes serve as weights and individual genotypes as predictors. Model parameters, including p-value thresholds and LD pruning criteria, are typically optimized via grid search in a validation dataset |
.png)
From a statistical modeling perspective, the C+T approach can be interpreted as a sparse estimation strategy that performs variable selection through hard thresholding. Under this framework, only variants exceeding a predefined significance threshold are retained, implicitly approximating the genetic architecture of a trait as being driven by a limited number of loci with relatively large effects. While this formulation simplifies the model structure, it also entails a substantial reduction of correlation information among variants. In practice, achieving a balance between model simplicity and predictive performance typically requires systematic exploration across a range of significance thresholds and linkage disequilibrium parameters, with model selection guided by validation data to identify an appropriate parameter configuration.
C+T offers advantages including simplicity, low computational cost, direct compatibility with GWAS summary statistics, and strong interpretability, making it a useful baseline method or rapid screening tool for large-scale traits (Wang et al., 2023). However, its “hard” LD pruning discards potentially informative variants and prevents optimal weighting within LD blocks. Moreover, its sensitivity to threshold selection and reference panels often leads to poor transferability across populations (Jayasinghe et al., 2024; Kachuri et al., 2024). Fundamentally, this reflects substantial shifts in the estimand when LD structure is ignored in cross-population settings.
The C+T approach is computationally efficient, interpretable, and compatible with GWAS summary statistics, making it a widely used baseline method. However, its reliance on hard LD pruning may discard informative variants and lead to suboptimal weighting within LD blocks. In addition, the method is sensitive to parameter choices and LD reference panels, which limits its portability across populations.
1.2 LD-aware and bayesian methods
To overcome the limitations of C+T in handling LD, methods that explicitly model LD structure have been developed, including LDpred, LDpred2, and PRS-CS (Weissbrod et al., 2022). These approaches incorporate an LD matrix ![]() from a reference panel and jointly estimate SNP effects via de-correlation and shrinkage, aiming to obtain the posterior expectation:
from a reference panel and jointly estimate SNP effects via de-correlation and shrinkage, aiming to obtain the posterior expectation:
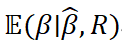
At the modeling level, different approaches distinguish themselves primarily through the choice of prior distributions imposed on effect sizes, thereby reflecting distinct assumptions about the underlying genetic architecture. For instance, the LDpred family of methods typically adopts a point-normal mixture prior to explicitly capture sparsity in effect sizes, whereas PRS-CS employs a continuous shrinkage prior that allows effect sizes to vary more smoothly across the genome and enables data-driven estimation of global hyperparameters (Sima et al., 2024). These differences in prior specification fundamentally represent alternative trade-offs between sparsity and continuity in modeling genetic effects.
From a unified statistical perspective, these methods can be expressed as:
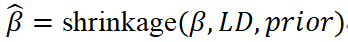
That is, regularized estimation of GWAS effects under LD constraints and prior assumptions. Compared to C+T, these approaches avoid discrete LD pruning and instead achieve near-optimal linear combinations within LD regions, typically resulting in improved predictive performance and greater parameter stability.
However, their performance depends critically on the consistency between the LD reference panel and the target population. When ancestry mismatch or structural variation exists, LD discrepancies may bias effect estimation and degrade prediction accuracy (Jayasinghe et al., 2024). In addition, large-scale LD matrix computation imposes substantial computational burden, and although methods such as LDpred2 have improved efficiency, trade-offs between accuracy and scalability remain.
1.3 Functional annotation and prior integration
To better approximate causal variants and reduce “tagging effects,” recent methodological advances have incorporated functional annotation information into effect estimation. Stratified LD score regression (S-LDSC) estimates heritability contributions across annotation categories within the baseline-LD framework, providing a basis for constructing informative priors (Sima et al., 2024).
Representative methods such as AnnoPred and LDpred-funct integrate functional annotations into Bayesian priors, assigning higher inclusion probabilities or weaker shrinkage to biologically relevant variants (e.g., eQTLs or open chromatin regions), thereby improving signal-to-noise ratio and predictive performance. Because some functional annotations are relatively conserved across populations, these approaches can partially mitigate cross-population bias induced by differences in LD structure and allele frequencies.
From a statistical modeling perspective, the incorporation of functional annotations can be viewed as integrating external biological information into the effect estimation process. This is typically achieved by imposing structured priors that differentiate across variant categories, thereby enabling a more structured representation of effect size distributions. As a result, the model no longer relies solely on data-driven estimation, but instead leverages prior information to guide shrinkage, improving both signal detection and estimation stability.
However, functional annotations are often tissue-and environment-specific and subject to measurement and annotation biases, which may lead to prior misspecification. Therefore, in practice, multi-tissue integration and small-sample recalibration in the target population are recommended to enhance robustness (Jayasinghe et al., 2024).
1.4 Multi-ancestry transfer and cross-population models
The primary objective of multi-ancestry PRS is to balance sample size and genetic heterogeneity across populations by jointly leveraging GWAS data from multiple ancestries and ancestry-specific LD structures, thereby improving generalization performance (Ruan et al., 2021; Zhang et al., 2023). For example, PRS-CSx performs continuous shrinkage separately within each ancestry and integrates results using shared hyperparameters and data-driven weights, capturing both shared and population-specific effects (Sima et al., 2024).
In addition, transfer learning and meta-GWAS approaches are widely used. Heterogeneity-aware meta-analysis and random-effects models can reduce estimation bias across studies, while stacking or reweighting methods can be applied when target population data are limited, enabling recalibration and adaptation of multiple ancestry-specific scores (Cai et al., 2021; Gunn et al., 2024).
Within a unified statistical framework, multi-ancestry approaches can be viewed as mechanisms for integrating and reconstructing the underlying estimands across populations. Rather than simply pooling data from multiple sources, these methods recharacterize the target estimand by balancing shared genetic effects against population-specific heterogeneity, typically through weighting schemes or hierarchical modeling. This allows the resulting predictive function to better accommodate cross-population variation.
The core challenge lies in accommodating differences in LD structure, allele frequency spectra, and effect heterogeneity across populations. In practice, this requires ancestry inference, local LD similarity assessment, and frequency-aware weighting of training data. Evaluation should include relative R²/AUC, calibration metrics, and decision-curve net benefit in the target population to comprehensively assess portability (Jung et al., 2025).
1.5 Relationship between PRS and SNP heritability: a unified view of variance explanation and individual prediction
Within the statistical genetics framework, PRS and SNP heritability are not independent quantities but rather complementary representations of the same underlying genetic information at different levels. SNP heritability is typically defined as the proportion of phenotypic variance explained by additive genetic effects captured by a given set of markers under specific model assumptions, representing a population-level variance decomposition. In contrast, PRS aggregates these effects into an individual-level predictive function that quantifies relative genetic risk.
Theoretically, the predictive performance of PRS is bounded by SNP heritability. Under ideal conditions-unbiased effect estimation, perfectly matched LD structure, and infinite sample size-PRS can approach the maximum variance explained by SNP heritability. In practice, however, PRS performance is typically lower due to estimation error, LD mismatch, and shrinkage-induced bias. Therefore, PRS performance should not be interpreted as a direct measure of trait heritability, but rather as an indicator of how effectively genetic signals can be identified and utilized under given data and model constraints.
This relationship can be formalized as a variance-prediction duality in statistical genetics (Fang, 2026): SNP heritability quantifies variance explained at the population level, whereas PRS reflects predictive ability at the individual level. The former corresponds to variance component estimation in random-effects models, while the latter corresponds to predictive functions constructed under shrinkage and regularization. Although both are theoretically linked through effect size distributions and LD structure, they often correspond to different estimands in practice due to differences in model assumptions and data structures.
This perspective is particularly important for understanding cross-population prediction. When allele frequency spectra, LD structure, or effect distributions differ across populations, both SNP heritability and PRS estimands may change, with PRS being more sensitive due to its dependence on specific effect estimates and LD modeling. Thus, the decline in cross-population predictive performance can be viewed as a manifestation of estimand mismatch at the level of individual prediction.
Based on this understanding, both PRS construction and evaluation should explicitly consider its relationship with SNP heritability. On the one hand, heritability estimates provide a theoretical upper bound and reference for PRS performance; on the other hand, improvements in effect estimation, ancestry-aware LD modeling, and target-specific recalibration are essential to narrow the gap between PRS performance and its theoretical limit, thereby enhancing predictive accuracy and cross-population portability.
2 Training-Validation-External Generalization Workflow
From a statistical inference perspective, the construction and evaluation of PRS/PGS should follow a staged process of estimation-regularization-prediction-validation, with the core objective of obtaining a stable and interpretable predictive function for the target population while avoiding overfitting. This workflow not only involves data partitioning and model selection, but also directly relates to the definition of the statistical target (estimand) and its consistency across different data domains.
2.1 Data splitting and internal validation
To ensure unbiased model development and evaluation, a three-stage framework is typically adopted: training, validation, and testing sets. The training set is used for effect estimation and model fitting; the validation set is used for hyperparameter tuning and recalibration (e.g., p-value thresholds, shrinkage strength, and global scaling factors); and the test set is reserved for one-time performance evaluation (Sima et al., 2024). This process corresponds to the “effect size → shrinkage → PRS” stage in the statistical inference pipeline and represents the key point at which model bias enters.
In both human genetics and crop breeding contexts, particular attention must be paid to sample structure dependence. Related individuals (e.g., families, lines, or close relatives) should be assigned to the same data split to avoid information leakage. At the same time, the distribution of phenotypes and key covariates (e.g., sex, batch effects, ancestry principal components) should be comparable across splits. For studies with limited sample sizes, nested cross-validation or leave-group-out strategies (e.g., by population, environment, or experimental site) can improve estimation stability and reduce selection bias (Lennon et al., 2024).
In the preprocessing stage, genotype-phenotype harmonization must be strictly reproducible, including alignment of reference alleles and genomic coordinates, removal of ambiguous variants, and the use of LD reference panels and allele frequency estimates matched to the target population. Different methods reflect distinct modeling assumptions in their tuning strategies: C+T relies on grid search over p-value thresholds and LD parameters, whereas LD-aware and Bayesian approaches adjust prior strength or LD block structure for shrinkage estimation (Wang et al., 2023; Sima et al., 2024). During validation, only linear recalibration (e.g., slope and intercept adjustment) should be permitted. Once the model is frozen, any form of re-tuning (“information leakage”) must be strictly avoided to ensure independence of testing and external evaluation.
2.2 External generalization and cross-population evaluation
External generalization is the core step in evaluating PRS portability. It is fundamentally a statistical inference problem under domain shift, assessing whether a predictive function estimated in the training data can maintain stable performance across different ancestries, environments, or technical platforms (Ruan et al., 2021). This stage corresponds to the “PRS → prediction” step and represents the primary point where cross-population failure occurs.
The standard workflow includes independent quality control and allele alignment for external datasets, selection of appropriate LD reference panels based on principal component analysis or ancestry inference, and computation of PRS on a consistent scale. Performance evaluation should then be conducted independently in the external dataset, including discrimination, calibration, and utility assessment, while documenting differences in phenotype definitions and measurement error (Kachuri et al., 2024). Numerous studies have shown that PRS trained in European populations typically experience a 40-60% reduction in predictive accuracy when applied to non-European populations. Multi-ancestry training or methods (e.g., PRS-CSx, CT-SLEB) can partially recover performance, in some cases reaching approximately 80% or more of the source population performance (Duncan et al., 2019; Jung et al., 2025).
From a statistical perspective, this performance decay can be understood as an estimand mismatch caused by differences in LD structure, allele frequency spectra, and effect size distributions across populations. To disentangle structural differences from environmental effects, it is recommended to design multiple external validation datasets (e.g., across ancestries, locations, or years) and perform systematic sensitivity analyses, such as replacing LD reference panels or excluding high-LD regions (Wang et al., 2023).
In addition, cross-population applications must account for data heterogeneity, including inconsistencies in phenotype definitions, missing environmental records, and platform-specific batch effects, all of which may amplify extrapolation error. Therefore, external validation design should incorporate standardized data processing, multidimensional data recording, and cross-platform harmonization to improve interpretability and reproducibility.
2.3 Performance Evaluation Metrics
The effectiveness of PRS/PGS models should be assessed using a multi-dimensional framework that captures predictive accuracy, calibration, and practical decision value. For binary outcomes (e.g., disease status), the area under the receiver operating characteristic curve (AUC) is a primary measure of discrimination (Lennon et al., 2024). For continuous traits, the proportion of variance explained (R²) and correlation coefficients (r) reflect predictive accuracy, and can be transformed to the liability scale (R²_liability) for cross-population comparability (Sima et al., 2024).
In terms of effect size, odds ratios (OR) or hazard ratios (HR) per standard deviation of PRS are commonly reported in clinical studies (Patel et al., 2023). Cross-population performance can be quantified using the transferability ratio  and differences in AUC (Duncan et al., 2019). From a statistical interpretation perspective, R² reflects variance explained by genetic signals, whereas AUC reflects the model’s ability to rank individuals, corresponding to “variance explanation” and “predictive discrimination,” respectively.
and differences in AUC (Duncan et al., 2019). From a statistical interpretation perspective, R² reflects variance explained by genetic signals, whereas AUC reflects the model’s ability to rank individuals, corresponding to “variance explanation” and “predictive discrimination,” respectively.
At the application level, threshold selection should be guided by decision context. In medical studies, decision curve analysis (DCA) can be used to determine clinically meaningful thresholds based on disease prevalence and intervention costs. For example, OR ≈ 1.5 with good calibration may support enhanced risk screening, whereas OR ≥ 2.0 may justify early intervention for high-risk individuals (Jung et al., 2025). In crop breeding, threshold optimization should consider heritability, environmental heterogeneity, and economic weights, and can be informed by simulations or multi-environment trial data.
To avoid over-reliance on single metrics, it is recommended to report uncertainty intervals, sensitivity analyses, and calibration curves, thereby providing a more robust and comprehensive evaluation of model performance.
3 Joint Prediction of PRS with Environmental and Lifestyle Factors
Within the framework of statistical genetics, PRS fundamentally captures an individual’s baseline genetic risk under given genetic information. However, in real-world systems, phenotypes are jointly determined by genetic effects and environmental exposures. Therefore, jointly modeling PRS with environmental or lifestyle factors can be interpreted as a conditional extension of the predictive functional, in which environmental dependence is introduced on top of genetic main effects, thereby improving both predictive accuracy and decision utility.
3.1 Modeling gene-environment interaction (G×E)
In multi-environment trials (METs) or longitudinal population studies, gene-environment interaction (G×E) determines the stability and transferability of predictive functions across different environmental conditions. By using PRS/PGS as a low-dimensional representation of genetic main effects and incorporating environmental variables (e.g., site, year, climate, or lifestyle exposures), a reaction norm framework can be constructed:
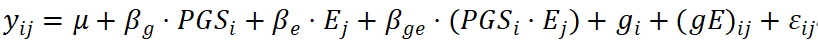
where gi and (ge)ij denote random genetic effects and their interaction with the environment, respectively. These can be modeled using factor-analytic (FA) or kernel-based covariance structures to capture cross-environment correlations. In high-dimensional environmental settings, dimensionality reduction techniques such as principal component analysis or ecological indices are typically applied to distinguish general adaptability from specific adaptability.
In human studies, environmental exposures are often time-dependent (e.g., smoking, diet, physical activity), and can be modeled using logistic regression or survival analysis:
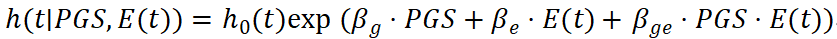
Within a statistical modeling framework, the incorporation of G×E effects extend the predictive function from one that depends solely on genetic information to one that is explicitly conditioned on environmental context. In this setting, PRS no longer represents a marginal genetic effect averaged across environments; rather, it acts as a modifier of environmental effects, allowing the relationship between environmental exposure and phenotype to vary systematically across different levels of genetic risk. From this perspective, gene-environment interaction can be understood as heterogeneity in environmental response patterns across strata of polygenic scores, whereby the functional form of the environment-phenotype relationship becomes dependent on PRS. Consequently, prediction shifts from a marginal formulation to a conditional, context-dependent representation (Figure 2).
|
Figure 2 Conditional prediction under gene-environment interaction (G×E): PRS as a function modifier Note: Illustration of gene-environment interaction (G×E) as differences in slopes across environmental gradients. Each line represents a quantile of polygenic scores (PGS), showing that the effect of environmental exposure on phenotype depends on genetic background. This reflects a conditional predictive function, where PRS modifies the relationship between environment and outcome (PRS = f(x) → f(x, E)) |
.png)
However, this process is susceptible to multiple sources of bias, including measurement error in environmental variables, time misalignment, and confounding induced by gene-environment correlation (rGE). Therefore, stratified analyses, instrumental variable approaches, or negative control designs are recommended, along with replication in independent cohorts to ensure robustness of interaction effects (Kachuri et al., 2024; Sima et al., 2024).
3.2 Statistical and machine learning models
Within traditional statistical frameworks, joint models are typically formulated as linear or generalized linear models incorporating genetic, environmental, and interaction effects:
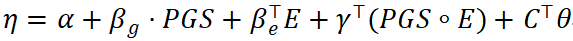
where C represents covariates such as age, sex, ancestry principal components, and batch effects.
In high-dimensional settings, to prevent overfitting, regularization methods such as ridge regression, elastic net, or group lasso are commonly used to impose hierarchical shrinkage on interaction terms, balancing model complexity and predictive stability. In MET designs, leave-group-out cross-validation (e.g., by site or year) should be employed to prevent environmental information leakage.
For time-varying exposures, piecewise time-varying coefficient models or joint longitudinal-survival models can be used to mitigate bias arising from temporal misalignment (Kachuri et al., 2024).
Machine learning methods provide complementary tools for capturing nonlinearities and higher-order interactions. Random forests and gradient boosting machines (GBM) are robust to data heterogeneity and missingness, while deep learning models are suitable for integrating multi-source phenotypic data (e.g., high-throughput phenotyping or wearable device data). However, from a statistical inference perspective, increased model flexibility often comes at the cost of higher estimation bias and greater uncertainty in generalization. Therefore, nested cross-validation, class imbalance reweighting, and post hoc calibration (e.g., Platt scaling or isotonic regression) are essential to control overfitting (Sima et al., 2024).
For model interpretability, SHAP values or permutation importance can be used to quantify the marginal contributions of PRS, key environmental variables, and their interactions. Stability selection across different ancestries or ecological populations can further help identify predictors with consistent effects.
3.3 Predictive gain and decision utility of joint models
The improvement in predictive performance achieved by joint modeling of PRS and environmental factors can be quantified from a variance decomposition perspective:
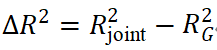
where 
 ,
, 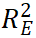
 , and
, and 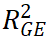 represent the contributions of genetic main effects, environmental main effects, and interaction effects, respectively, and
represent the contributions of genetic main effects, environmental main effects, and interaction effects, respectively, and 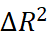 reflects the incremental gain of the joint model over PRS alone.
reflects the incremental gain of the joint model over PRS alone.
The magnitude of this gain depends on the variability of environmental factors and the strength of G×E interactions. When environmental exposures are modifiable and widely distributed in the target population, joint models often produce steeper risk gradients in the high-risk tail, thereby increasing net benefit in decision curve analysis (Kachuri et al., 2024; Sima et al., 2024).
In population-based studies, the evaluation of PRS is more appropriately conducted within a comparative modeling framework rather than relying on a single model specification. A baseline model incorporating key covariates, such as age, sex, and ancestry principal components, is typically used as a reference, upon which PRS, environmental exposures, and their interaction terms are incorporated. Model performance can then be assessed in a multidimensional manner, capturing both discrimination and explanatory capacity, using metrics such as AUC, R² (or liability-scale R²), effect sizes per standard deviation of PRS (e.g., OR or HR), and net benefit derived from decision curve analysis. Furthermore, stratifying individuals according to lifestyle or environmental exposure levels allows for a more nuanced characterization of risk distributions and calibration patterns, thereby facilitating the evaluation of the “risk equivalence” phenomenon-namely, the extent to which adverse environmental exposures may attenuate or offset the protective effects associated with lower genetic risk (Sima et al., 2024).
In crop breeding, a two-stage decision framework can be implemented: early-generation selection using PGS to improve selection accuracy (correlation r), followed by estimation of environment-specific interaction effects (βae) in small samples from target environments. This enables a strategy of “general adaptability selection + environment-specific optimization,” with theoretical genetic gain expressed as:
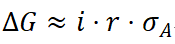
where i denotes selection intensity and 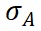 the additive genetic standard deviation (Ruan et al., 2021). This framework demonstrates that integrating PRS with environmental information not only improves predictive accuracy but also translates directly into higher selection efficiency and practical utility.
the additive genetic standard deviation (Ruan et al., 2021). This framework demonstrates that integrating PRS with environmental information not only improves predictive accuracy but also translates directly into higher selection efficiency and practical utility.
4 Ethical and Population Fairness Issues
In the practical application of polygenic risk scores (PRS/PGS), ethical and population fairness concerns arise not only from social structural differences but are also deeply rooted in differences in the applicability of statistical models across populations. From a statistical genetics perspective, cross-population inequity can be understood as inconsistency in the statistical target (estimand) represented by the predictive function across different data domains, leading to systematic performance bias and decision risk.
4.1 Inequity in cross-population prediction
A large body of evidence indicates that PRS/PGS trained predominantly on European populations exhibit substantially reduced predictive performance in non-European populations (approximately 40-80% decline). This reduction is primarily driven by differences in allele frequency spectra and linkage disequilibrium (LD) structure, sampling bias in GWAS discovery and effect estimation, and inconsistencies in phenotype definition and measurement. These factors collectively result in decreased explanatory power, calibration bias, and unstable decision thresholds during extrapolation, thereby increasing misclassification risk and leading to inequitable allocation of resources (Duncan et al., 2019; Martin et al., 2019; Zhang et al., 2023).
From a statistical inference perspective, this phenomenon can be understood as arising from systematic differences across populations in linkage disequilibrium structure, allele frequency spectra, and effect size distributions, which lead to a misalignment between the estimand defined in the training data and the target of prediction in the external population-commonly referred to as estimand mismatch.
Among different populations, individuals of African ancestry and admixed populations are particularly affected, largely due to their higher genetic diversity and the relative lack of representative LD reference panels and functional annotation resources. As a result, tag SNPs are less able to reliably proxy underlying causal variants (Ding et al., 2023; Kachuri et al., 2024).
Similar issues are observed in crop breeding systems. Differences in subspecies structure, ecological population stratification, and the target population of environments (TPE) can lead to failure of PGS models trained on elite germplasm or specific environments when applied to local varieties or marginal ecological conditions. Gene-environment interactions (G×E) further amplify these discrepancies, reducing selection efficiency under environmental extrapolation and potentially leading to systematic neglect of important genetic signals relevant to smallholder farming systems or marginal environments, thereby creating forms of “hidden inequity” (Sima et al., 2024).
4.2 Ethical and societal considerations
Overinterpretation of PRS as a deterministic measure of an individual’s “genetic destiny” may lead to distorted risk perception, stigmatization, and reinforcement of social stereotypes. When predictive performance and decision thresholds are not comparable across populations, the direct application of PRS in screening or intervention may result in unequal distribution of healthcare resources, thereby exacerbating existing health disparities (Martin et al., 2019; Lewis and Green, 2021; Andreoli et al., 2024). In addition, issues related to data governance and privacy are critical, including risks of re-identification, policies for returning results to participants, and mechanisms for dynamic informed consent. These considerations require the integration of ethical evaluation throughout the entire research and implementation lifecycle (Adeyemo et al., 2021). In sensitive domains such as psychiatric traits, particular caution is needed in interpretation and communication to avoid reinforcing genetic determinism or misleading the public (Murray et al., 2020; Chapman, 2022).
In agricultural and breeding contexts, ethical challenges are more closely associated with structural imbalances in technology deployment. For example, recommendations based solely on large-scale data from a single environment may disadvantage niche ecological systems or smallholder farmers, and may, over time, reduce genetic diversity and compromise the resilience of food systems (Sima et al., 2024). Furthermore, access to and utilization of international germplasm resources and genomic data are often highly unequal. Without appropriate frameworks for intellectual property and benefit-sharing, such imbalances may exacerbate global disparities between developed and developing regions. Differences in farmers’ access to data and technology further constrain the equitable implementation of PRS/PGS in real-world agricultural systems.
4.3 Mitigation strategies: a multi-layer framework from data to governance
Addressing inequity in cross-population PRS/PGS applications requires systematic improvements at three levels: data, methodology, and governance.
At the data level, efforts should focus on expanding multi-ancestry and multi-ecological GWAS datasets and reference panels to improve coverage of low-frequency and structural variants, as well as developing comprehensive functional annotation resources across tissues and environments. Standardization of data quality control (QC), genomic coordinates, and allele coding conventions is essential to reduce technical heterogeneity across studies (Zhang et al., 2023; Kachuri et al., 2024; Kullo, 2024). In addition, promoting cross-institutional data sharing and establishing standardized consortia, along with dynamically updated public resource repositories, will improve both representativeness and accessibility of data.
At the methodological level, advances are needed in multi-ancestry joint modeling, hierarchical modeling, ancestry-aware LD modeling, and transfer learning approaches (e.g., PRS-CSx, CT-SLEB, Joint-Lassosum). Incorporating local ancestry information and domain adaptation techniques can further enhance model adaptability to population structure differences. Small-sample reweighting or recalibration in the target population has been shown to effectively reduce performance gaps in practice (Zhang et al., 2023; Kachuri et al., 2024). Furthermore, integrating functional annotations and causal inference approaches as biologically informed priors can reduce noise and improve robustness in cross-population prediction.
At the evaluation and governance level, it is essential to establish a systematic “fairness metric framework,” reporting performance metrics separately across populations, including AUC, R², calibration slope and intercept, Brier score, net benefit, and reclassification metrics (NRI). Performance differences should be quantified using measures such as the transferability ratio (T) and ΔAUC. In addition, decision thresholds and rules should be optimized for each population, and uncertainty measures (e.g., confidence intervals and calibration curves) should be reported to avoid overinterpretation of single metrics. Adoption of transparent reporting standards (e.g., Polygenic Score Catalog, PRS Reporting Statement) and regulatory and ethical compliance frameworks (Wand et al., 2020; Lewis et al., 2024; Xiang et al., 2024) will further ensure that PRS/PGS applications are scientifically robust, interpretable, and equitable.
5 Discussion
From a unified statistical inference framework, the performance differences among existing PRS/PGS methods fundamentally arise from distinct modeling assumptions regarding effect size distributions, linkage disequilibrium (LD) structure, and sparsity, thereby corresponding to different statistical targets (estimands). From this perspective, methodological evolution can be viewed as a progressive approximation to the question of how GWAS-derived effects can be transformed into stable predictive functions.
Compared with baseline approaches such as clumping and thresholding (C+T), LD-aware Bayesian shrinkage methods (e.g., LDpred2 and PRS-CS) apply continuous shrinkage to effect sizes under LD constraints, typically achieving higher predictive performance and smoother behavior with respect to hyperparameters under the same training data and reference panels. When functional annotations are incorporated (e.g., LDpred-funct and AnnoPred), differential shrinkage or preferential inclusion of variants in functionally enriched regions can further improve the signal-to-noise ratio and partially mitigate cross-population bias induced by tagging effects (Sima et al., 2024). In settings involving heterogeneous data or extrapolation, multi-ancestry and cross-population approaches (e.g., PRS-CSx, hierarchical modeling, and model stacking) balance shared and population-specific effects, often achieving a better trade-off between transferability and stability (Zhang et al., 2023).
Based on these insights, a practical three-step decision framework can be adopted, centered on “scenario-resource-validation.” First, modeling strategies should be selected based on the sample size and representativeness of the target population (population-specific models versus multi-ancestry transfer models). Second, method selection should consider the availability of LD reference panels and functional annotations (annotation-informed Bayesian methods versus LD-aware baseline approaches). Third, model selection should be driven by external validation, comparing relative R²/AUC, calibration slope, and decision-curve net benefit in held-out or independent datasets, with the final model applied only after being frozen (Kachuri et al., 2024).
At the data and modeling levels, future improvements in PRS performance require both expanded data coverage and advances in statistical methodology. On the one hand, efforts should focus on increasing the representation of multi-ancestry GWAS and LD reference panels, improving the capture of low-frequency and structural variants, and implementing standardized quality control across centers and platforms in human studies, as well as covering the target population of environments (TPE) through multi-environment trials (METs) in crop systems (Wang et al., 2018). On the other hand, modeling strategies should incorporate ancestry-aware LD structures, hierarchical random effects, and transfer learning approaches to decompose shared and population-specific effects. In target populations, recalibration (e.g., slope and intercept adjustment) and reweighting (stacking) can reduce predictive bias, while the integration of local ancestry and functional annotations can further improve cross-population robustness (Cai et al., 2021; Zhang et al., 2023).
At the evaluation level, a shift from single performance metrics to a systematic assessment framework is needed. In addition to reporting R² and AUC, studies should report the transferability ratio (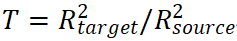 ), population-stratified calibration metrics (slope and intercept), and decision-curve net benefit. Sensitivity analyses with respect to LD reference panels, functional annotation strength, and model parameters should also be conducted, forming a closed-loop evaluation framework of “training-extrapolation-recalibration-re-evaluation.”
), population-stratified calibration metrics (slope and intercept), and decision-curve net benefit. Sensitivity analyses with respect to LD reference panels, functional annotation strength, and model parameters should also be conducted, forming a closed-loop evaluation framework of “training-extrapolation-recalibration-re-evaluation.”
At the application level, PRS/PGS are evolving from predictive tools toward decision-support systems. In medicine, combining PGS with age, family history, biomarkers, and lifestyle factors enables stratified screening and personalized interventions for high-burden diseases such as cardiovascular, metabolic, and certain cancers. In crop breeding, PGS is methodologically aligned with genomic selection (GS), and can be used for early-stage preselection and multi-environment reaction norm modeling to achieve coordinated optimization of “general adaptability and environment-specific selection,” thereby substantially improving genetic gain per unit time in complex stress-related traits (Wang et al., 2018; Alemu et al., 2024).
Despite these advances, several key bottlenecks remain. First, imbalances in multi-ancestry data and functional annotation resources lead to training bias and uncertainty in evaluation. Second, low-frequency and rare variants, as well as structural variants (e.g., SVs and CNVs), are not fully captured under current “tag SNP” frameworks, requiring advances in pangenome references, long-read sequencing, and multi-omics data integration for improved causal inference. Third, cross-platform batch effects and residual population structure may further amplify extrapolation errors (Du et al., 2025). From an ethical and governance perspective, medical applications must address risks of genetic determinism and discrimination, and establish frameworks for dynamic consent and data sovereignty; in breeding, considerations of biodiversity conservation, equitable benefit sharing, and support for smallholder systems are essential to avoid structural bias driven by performance optimization alone (Gorjanc et al., 2017; Broesch et al., 2020).
Looking forward, the development of PRS/PGS can be summarized within an integrated paradigm: multi-ancestry data expansion + causal and functional annotation integration + ancestry-aware modeling + environment coupling + recalibration and fairness evaluation.
This paradigm will facilitate the transition of PRS/PGS from standalone predictive tools to comprehensive platforms that are transferable, interpretable, and governable.
6 Conclusion
Polygenic risk scores (PRS/PGS) systematically integrate GWAS-derived effect estimates to transform locus-trait associations into actionable individual-level predictive measures. In both human medicine and crop breeding, they support risk stratification, early screening, and selection decisions, serving as a key bridge between fundamental genetics and translational applications.
From a statistical genetics perspective, PRS is not merely a predictive tool but a model-dependent predictive function, whose performance and scope are jointly determined by training data, LD structure, and effect estimation methods. In correspondence with SNP heritability as a measure of variance explained, PRS represents the expression of genetic signal at the individual prediction level. Together, they form a variance-prediction duality. Within this framework, the decline in cross-population predictive performance can be understood as an estimand mismatch arising from differences in LD structure, allele frequency spectra, and effect distributions across populations.
At present, cross-population robustness remains the central bottleneck for translating PRS/PGS into clinical and agricultural practice. Advances in multi-ancestry data resources and methodology are progressively addressing this challenge. Multi-ancestry training, hierarchical modeling, ancestry-aware LD modeling, and the integration of local ancestry information enable improved balance between shared and population-specific effects. The incorporation of functional annotation and causal refinement further reduces the impact of tagging effects on cross-population prediction. At the application level, adherence to a standardized workflow- “training-validation-freezing-external evaluation” -together with multi-dimensional performance metrics for predictive accuracy and fairness, is increasingly recognized as best practice.
Looking forward, the development of PRS/PGS is expected to follow three major directions. First, integration of causal inference and functional annotation will enhance signal identification and cross-population robustness through structured priors. Second, multi-ancestry modeling and transfer learning will reduce performance gaps in underrepresented populations. Third, coupling with environmental and lifestyle factors will extend predictive functions into context-dependent models through explicit modeling of gene-environment interaction. In medicine, these advances will support stratified screening and personalized interventions for high-burden diseases; in crop breeding, they will enable efficient strategies combining general adaptability with environment-specific optimization.
At the same time, several key challenges remain. These include imbalances in multi-ancestry data and annotation resources, limited coverage of low-frequency and structural variants, cross-platform batch effects, and residual population structure, as well as ethical and governance considerations such as data sovereignty and fairness. With the development of pangenome reference systems, long-read sequencing, and multi-omics integration, along with the establishment of open benchmarks, standardized quality control, and transparent reporting frameworks, PRS/PGS are expected to evolve into a general predictive platform that is scientifically robust and socially responsible, with broad applications in global health and food security.
Author Contributions
Xuanjun Fang conducted the study, including literature review, data analysis, and drafting and revising the manuscript. The author has read and approved the final version of the manuscript.
Acknowledgements
This work was supported by the Major Program of the National Natural Science Foundation of China (Grant No. 30490254).
Adeyemo A., Balaconis M., Darnes D., and Zhou A., 2021, Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps, Nature Medicine, 27(11): 1876-1884.
https://doi.org/10.1038/s41591-021-01549-6
Alemu A., Åstrand J., Montesinos-López O., Sánchez J., Fernández-González J., Tadesse W., Vetukuri R., Carlsson A., Ceplitis A., Crossa J., Ortiz R., and Chawade A., 2024, Genomic selection in plant breeding: Key factors shaping two decades of progress, Molecular Plant, 17(4): 552-578.
https://doi.org/10.1016/j.molp.2024.03.007
Andreoli L., Peeters H., Van Steen K., and Dierickx K., 2024, Taking the risk: Ethical reasons and moral arguments in the clinical use of polygenic risk scores, American Journal of Medical Genetics Part A, 194(7): 1939-1956.
https://doi.org/10.1002/ajmg.a.63584
Broesch T., Crittenden A., Beheim B., Blackwell A., Bunce J., Colleran H., Hagel K., Kline M., Mcelreath R., Nelson R., Pisor A., Prall S., Pretelli I., Purzycki B., Quinn E., Ross C., Scelza B., Starkweather K., Stieglitz J., and Mulder M., 2020, Navigating cross-cultural research: Methodological and ethical considerations, Proceedings of the Royal Society B, 287(1935): 20201245.
https://doi.org/10.1098/rspb.2020.1245
Cai M., Xiao J., Zhang S., Wan X., Zhao H., Chen G., and Yang C., 2021, A unified framework for cross-population trait prediction by leveraging the genetic correlation of polygenic traits, American Journal of Human Genetics, 108(4): 632-655.
https://doi.org/10.1016/j.ajhg.2021.03.002
Chapman C., 2022, Ethical, legal, and social implications of genetic risk prediction for multifactorial disease, Journal of Community Genetics, 14(5): 441-452.
https://doi.org/10.1007/s12687-022-00625-9
Ding Y., Hou K., Xu Z., Pimplaskar A., Petter E., Boulier K., Privé F., Vilhjálmsson B., Loohuis L., and Pasaniuc B., 2023, Polygenic scoring accuracy varies across the genetic ancestry continuum, Nature, 618(7966): 774-781.
https://doi.org/10.1038/s41586-023-06079-4
Du Z., He J., and Jiao W., 2025, Plant graph-based pangenomics: Techniques, applications, and challenges, aBIOTECH, 2025: 1-16.
https://doi.org/10.1007/s42994-025-00206-7
Duncan L., Shen H., Gelaye B., Meijsen J., Ressler K., Feldman M., Peterson R., and Domingue B., 2019, Analysis of polygenic risk score usage and performance in diverse human populations, Nature Communications, 10(1): 3328.
https://doi.org/10.1038/s41467-019-11112-0
Fang X.J., 2026, Genome-wide relationship matrix-based heritability estimation: statistical interpretation, comparability, and practical diagnostics in the GCTA-GREML framework, Computational Molecular Biology, 16(1): 11-20.
Fang X.J., and Wu W.R., 2026, Evolution of statistical genetic paradigms: from linkage analysis and candidate gene strategies to GWAS, Molecular Plant Breeding, 24(9): 2817-2829.
Gorjanc G., Gaynor R., and Hickey J., 2017, Optimal cross selection for long-term genetic gain, Theoretical and Applied Genetics, 131(9): 1953-1966.
https://doi.org/10.1007/s00122-018-3125-3
Gunn S., Wang X., Posner D., Cho K., Huffman J., Gaziano M., Wilson P., Sun Y., Peloso G., and Lunetta K., 2024, Comparison of methods for building polygenic scores for diverse populations, Human Genetics and Genomics Advances, 6: 100355.
https://doi.org/10.1016/j.xhgg.2024.100355
Hickey J., Chiurugwi T., Mackay I., and Powell W., 2017, Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery, Nature Genetics, 49(9): 1297-1303.
https://doi.org/10.1038/ng.3920
Jayasinghe D., Eshetie S., Beckmann K., Benyamin B., and Lee S., 2024, Advancements and limitations in polygenic risk score methods for genomic prediction: A scoping review, Human Genetics, 143(12): 1401-1431.
https://doi.org/10.1007/s00439-024-02716-8
Jung H.U., Jung H., Baek E., Kang J., Kwon S., You J., Lim J., and Oh B., 2025, Assessment of polygenic risk score performance in East Asian populations for ten common diseases, Communications Biology, 8(1): 374.
https://doi.org/10.1038/s42003-025-07767-9
Kachuri L., Chatterjee N., Hirbo J., Schaid D., Martin I., Kullo I., Kenny E., Pasaniuc B., Witte J., and Ge T., 2024, Principles and methods for transferring polygenic risk scores across global populations, Nature Reviews Genetics, 25(1): 8-25.
https://doi.org/10.1038/s41576-023-00637-2
Kullo I., 2024, Promoting equity in polygenic risk assessment through global collaboration, Nature Genetics, 56(9): 1780-1787.
https://doi.org/10.1038/s41588-024-01843-2
Lennon N., Kottyan L., Kachulis C., Abul-Husn N., Arias J., Belbin G., Below J., Berndt S., Chung W., Cimino J., and Kenny E., 2024, Selection, optimization and validation of ten chronic disease polygenic risk scores for clinical implementation in diverse US populations, Nature Medicine, 30(2): 480-487.
https://doi.org/10.1038/s41591-024-02796-z
Lewis A., and Green R., 2021, Polygenic risk scores in the clinic: New perspectives on familiar ethical issues, Genome Medicine, 13(1): 14.
https://doi.org/10.1186/s13073-021-00829-7
Lewis A., Chisholm R., Connolly J., Esplin E., Glessner J., Gordon A., Green R., Hakonarson H., Harr M., Holm I., Jarvik G., Karlson B., Kenny E., Kottyan L., Lennon N., Linder J., Luo Y., Martin L., Perez E., Puckelwartz M., Rasmussen-Torvik L., Sabatello M., Sharp R., Smoller J., Sterling R., Terek S., Wei W., and Fullerton S., 2024, Managing differential performance of polygenic risk scores across groups: Real-world experience of the eMERGE Network, American Journal of Human Genetics, 111(6): 999-1005.
https://doi.org/10.1016/j.ajhg.2024.04.005
Martin A., Kanai M., Kamatani Y., Okada Y., Neale B.M., and Daly M., 2019, Clinical use of current polygenic risk scores may exacerbate health disparities, Nature Genetics, 51(4): 584-591.
https://doi.org/10.1038/s41588-019-0379-x
Murray G., Lin T., Austin J., Mcgrath J., Hickie I., and Wray N., 2020, Could polygenic risk scores be useful in psychiatry? A review, JAMA Psychiatry, 78(2): 210-219.
https://doi.org/10.1001/jamapsychiatry.2020.3042
Patel A., Wang M., Ruan Y., Koyama S., Clarke S., Yang X., Tcheandjieu C., Agrawal S., Fahed A., Ellinor P., and Khera A., 2023, A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease, Nature Medicine, 29(8): 1793-1803.
https://doi.org/10.1038/s41591-023-02429-x
Ruan Y., Lin Y., Feng Y., Chen C., Lam M., Guo Z., He L., Sawa A., Martin A., Qin S., Huang H., and Ge T., 2021, Improving polygenic prediction in ancestrally diverse populations, Nature Genetics, 54(4): 573-580.
https://doi.org/10.1038/s41588-022-01054-7
Sima C., Step K., Swart Y., Schurz H., Uren C., and Möller M., 2024, Methodologies underpinning polygenic risk score estimation: A comprehensive overview, Human Genetics, 143(11): 1265-1280.
https://doi.org/10.1007/s00439-024-02710-0
Wand H., Lambert S., Tamburro C., Iacocca M., O'Sullivan J., Sillari C., Kullo I., Rowley R., Dron J., Dron J., Brockman D., Venner E., McCarthy M., Antoniou A., Easton D., Hegele R., Khera A., Chatterjee N., Kooperberg C., Edwards K., Vlessis K., Kinnear K., Danesh J., Parkinson H., Ramos E., Roberts M., Ormond K., Khoury M., Janssens A., Goddard K., Kraft P., MacArthur J., Inouye M., and Wojcik G., 2020, Improving reporting standards for polygenic scores in risk prediction studies, Nature, 591(7848): 211-219.
https://doi.org/10.1038/s41586-021-03243-6
Wang X., Xu Y., Hu Z., and Xu C., 2018, Genomic selection methods for crop improvement, The Crop Journal, 6(4): 330-340.
https://doi.org/10.1016/j.cj.2018.03.001
Wang Y., Namba S., Lopera E., Kerminen S., Tsuo K., Läll K., Kanai M., Zhou W., Wu K., Favé M., and Neale B., 2023, Global biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts, Cell Genomics, 3(1): 100241.
https://doi.org/10.1016/j.xgen.2022.100241
Weissbrod O., Kanai M., Shi H., Gazal S., Peyrot W., Khera A., Okada Y., Matsuda K., Yamanashi Y., Furukawa Y., and Price A., 2022, Leveraging fine-mapping and multi-population training data to improve cross-population polygenic risk scores, Nature Genetics, 54(4): 450-458.
https://doi.org/10.1038/s41588-022-01036-9
Xiang R., Kelemen M., Xu Y., Harris L., Parkinson H., Inouye M., and Lambert S., 2024, Recent advances in polygenic scores: Translation, equitability, methods and FAIR tools, Genome Medicine, 16(1): 33.
https://doi.org/10.1186/s13073-024-01304-9
Zhang H., Zhan J., Jin J., Zhang J., Lu W., Zhao R., Ahearn T., Yu Z., O’Connell J., Jiang Y., and Chatterjee N., 2023, A new method for multiancestry polygenic prediction improves performance across diverse populations, Nature Genetics, 55(10): 1757-1768.
https://doi.org/10.1038/s41588-023-01501-z




. FPDF(win)
. FPDF(mac)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. Xuanjun Fang
Related articles
. Polygenic risk scores
. Statistical genetics
. SNP heritability
. Cross-population prediction
. Linkage disequilibrium
. Bayesian shrinkage
. Transfer learning
. Gene-environment interaction
. Fairness
Tools
. Post a comment